Computing Initial and Terminal States¶
Preliminaries¶
In this tutorial, you will learn how to use the GPCCA to do the following:
compute macrostates of cellular dynamics.
visualize a coarse-grained transition matrix among macrostates.
classify macrostates as initial, terminal or intermediate.
The GPCCA may be combined with any CellRank kernel used to compute a cell-cell transition matrix, including the VelocityKernel, the PseudotimeKernel and the CytoTRACEKernel. We explain the kernel/estimator paradigm in more detail in our Getting Started with CellRank tutorial.
This tutorial notebook can be downloaded using the following link.
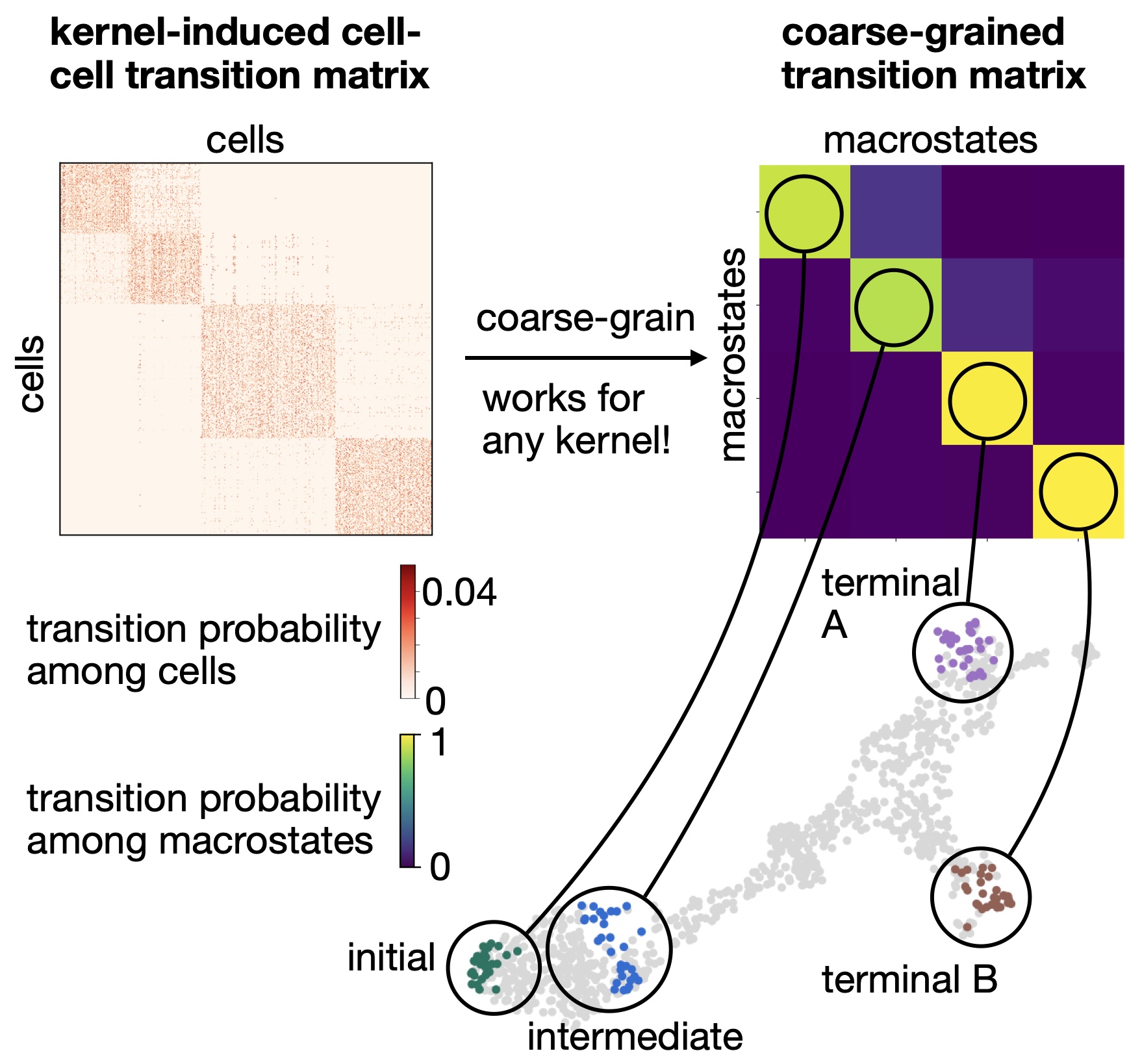
CellRank decomposes cellular dynamics into macrostates: Based on Generalized Perron Cluster Cluster analysis (GPCCA), CellRank identifies macrostates of cellular dynamics, which include initial, intermediate, and terminal states [Lange et al., 2022, Reuter et al., 2019, Reuter et al., 2018].¶
Note
If you want to run this on your own data, you will need:
a scRNA-seq dataset for which you computed a cell-cell transition matrix using any CellRank
kernel. For each kernel, there exist individual tutorials that teach you how to use them. For example, we have tutorials about using RNA velocity, experimental time points, and pseudotime, among others, to derive cell-cell transition probabilities.
Note
If you encounter any bugs in the code, our if you have suggestions for new features, please open an issue. If you have a general question or something you would like to discuss with us, please post on the scverse discourse.
Import packages & data¶
%load_ext autoreload
%autoreload 2
import sys
if "google.colab" in sys.modules:
!pip install -q git+https://github.com/scverse/cellrank
import cellrank as cr
import matplotlib.pyplot as plt
import scanpy as sc
import session_info2
cr.settings.logging_level = "INFO"
sc.set_figure_params(
frameon=False,
dpi=100,
)
import warnings
warnings.simplefilter("ignore", category=UserWarning)
To demonstrate the appproach in this tutorial, we will use a scRNA-seq dataset of pancreas development at embyonic day E15.5 [Bastidas-Ponce et al., 2019]. We used this dataset in our CellRank meets RNA velocity tutorial to compute a cell-cell transition matrix using the VelocityKernel; this tutorial builds on the transition matrix computed there. The dataset including the transition matrix can be conveniently acessed through cellrank.datasets.pancreas [Bastidas-Ponce et al., 2019, Bergen et al., 2020].
adata = cr.datasets.pancreas(kind="preprocessed-kernel")
vk = cr.kernels.VelocityKernel.from_adata(adata, key="T_fwd")
vk
VelocityKernel[n=2531, model='deterministic', similarity='correlation', softmax_scale=4.013]
Note
The method from_adata() allows you to reconstruct a CellRank kernel from an AnnData object. When you use a kernel to compute a transition matrix, this matrix, as well as a few informations about the computation, are written to the underlying anndata.AnnData object upon calling write_to_adata(). This makes it easy to write and read kernels from disk via the associated AnnData object.
As a reminder, below is a UMAP with cluster labels of this dataset [Bastidas-Ponce et al., 2019, Becht et al., 2019, McInnes et al., 2018]. In this tutorial, we will automatically identify the initial and terminal states.
sc.pl.embedding(adata, basis="umap", color="clusters")

Identify initial and terminal states¶
Initialize an estimator¶
The estimators allow you to analyze cell-state dynamics in CellRank. We initialize the GPCCA estimator here by passing the VelocityKernel [Reuter et al., 2019, Reuter et al., 2018]. However, the GPCCA estimator works with every CellRank kernel.
g = cr.estimators.GPCCA(vk)
print(g)
GPCCA[kernel=VelocityKernel[n=2531], initial_states=None, terminal_states=None]
We use the GPCCA estimator below to compute initial & terminal states; it may further be used to compute fate probabilities as we explain in the fate probabilities & driver genes tutorial.
Compute macrostates¶
Coarse-graining with GPCCA: Using the GPCCA algorithm, CellRank coarse-grains a cell-cell transition matrix onto the macro-state level. We obtain two key outputs: the soft assignment of cells to macrostates, and a matrix of transition probabilities among these macrostates [Reuter et al., 2019, Reuter et al., 2022, Reuter et al., 2018].¶
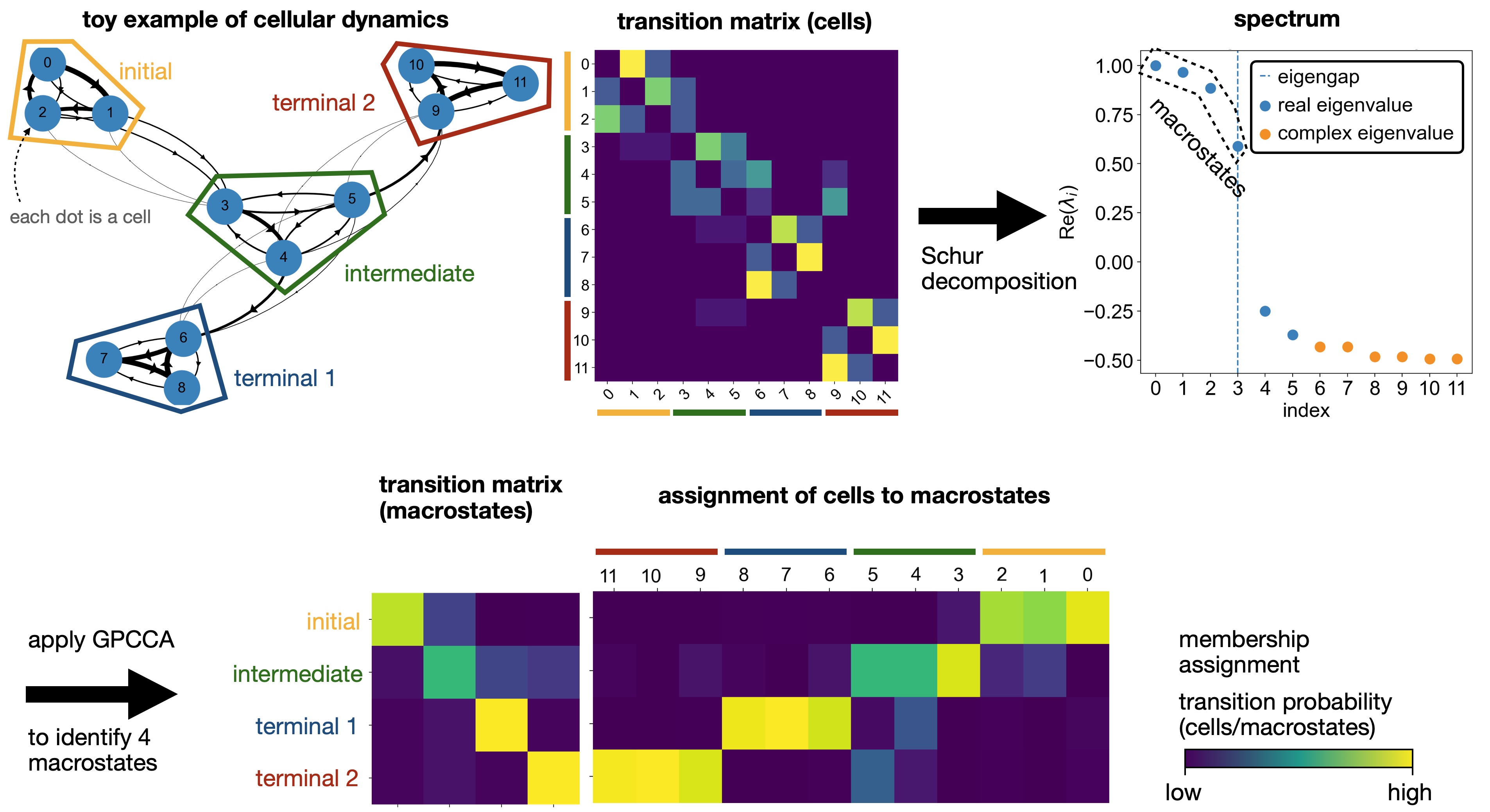
The computation of macrostates is based on the real Schur decomposition - essentially a generalization of the eigendecomposition. For non-symmetric transition matrices, as in our case, the eigendecomposition in general yields complex eigenvalues and eigenvectors, which are difficult to interpret. Thus, we revert to the real Schur decomposition [Reuter et al., 2019, Reuter et al., 2018].
Below, we also plot the real part of the top eigenvalues, sorted by their real part. Blue and orange denote real and complex eigenvalues, respectively. For real matrices, complex eigenvalues always appear in pairs of complex conjugates.
g.compute_schur()
g.plot_spectrum(real_only=True)
INFO Computing Schur decomposition
INFO When computing macrostates, choose a number of states NOT in ``
INFO Adding `adata.uns['eigendecomposition_fwd']`
`.schur_vectors`
`.schur_matrix`
`.eigendecomposition`
Finish (0.28s)

Using the real Schur decomposition, we compute macrostates with the GPCCA algorithm as implemented in pyGPCCA by maximizing for metastability [Reuter et al., 2019, Reuter et al., 2022, Reuter et al., 2018]. macrostates may contain initial, terminal and intermediate states of cellular dynamics.
In the plot above, the estimator automatically suggested a number of states to use (dashed vertical line); however, we will ignore that and compute a few more states to have a chance at identifying the Delta cell population.
Note
Make sure to have the scientific computing libraries PETSc and SLEPc installed for maximum performance, using e.g. conda install -c conda-forge petsc4py slepc4py. On most machines, this is strongly recommended if your datasets consists of over 10k cells. See our installation instructions.
g.compute_macrostates(n_states=11, cluster_key="clusters")
g.plot_macrostates(which="all", legend_loc="right", s=100)
INFO Computing 11 macrostates
INFO Adding `.macrostates`
`.macrostates_memberships`
`.coarse_T`
`.coarse_initial_distribution
`.coarse_stationary_distribution`
`.schur_vectors`
`.schur_matrix`
`.eigendecomposition`
Finish (3.56s)

We now have macrostates for Alpha, Beta, Epsilon and Delta populations, besides a few progenitor populations. We assign one label per macrostate based on the underlying 'clusters' annotation. However, that does not imply that all cells within one macrostate are from the same underlying cluster as we use majority voting. We can visualize the relationship between clusters and macrostates. We show below the distribution over cluster membership for each of the cells most confidently assigned to a certain macrostate.
g.plot_macrostate_composition(key="clusters", figsize=(7, 4))

With some exceptions, most macrostates recruit their top-cells from a single underlying cluster. This plotting function works for any observation-level covariate and can be useful when we have experimental time labels as we expect more mature states to come from later time points. See the RealTimeKernel and the corresponding tutorial to learn how CellRank makes use of experimental time points.
To get an idea of how these macrostates are related, we plot the coarse-grained transition matrix.
g.plot_coarse_T(annotate=False)

By default, macrostates are ordered according to their diagonal value, increasing from left to right. The diagonal value is a proxy for a states’ metastability, i.e. the probability of leaving the state once entered. While the Epsilon, Alpha and Beta states have high diagonal values, Delta cells have a low value because they are predicted to transition into Beta cells (check the corresponding matrix element).
Classify macrostates¶
Let’s try automatic terminal state identification first.
g.predict_terminal_states()
g.plot_macrostates(which="terminal", legend_loc="right", s=100)
INFO Adding `adata.obs['term_states_fwd']`
`adata.obs['term_states_fwd_probs']`
`.terminal_states`
`.terminal_states_probabilities`
`.terminal_states_memberships
Finish`

We can visually confirm our terminal state identification by comparing with well-known mature cell-type markers Ins1 and Ins2 for Beta cells, Gcg for Alpha cells and Ghrl for epsilon cells.
sc.pl.embedding(
adata,
basis="umap",
color=["Ins1", "Ins2", "Gcg", "Ghrl", "Hhex"],
ncols=5,
size=50,
)

We correctly identified some hormone-producing terminal cell states. However, we did not catch the Delta cells (marked by Hhex)! If we want to compute fate probabilities towards them later on, we need to annotate them as a terminal state. Luckily, this can be done semi-automatically as well:
g.set_terminal_states(states=["Alpha", "Beta", "Epsilon", "Delta"])
g.plot_macrostates(which="terminal", legend_loc="right", s=100)
INFO Adding `adata.obs['term_states_fwd']`
`adata.obs['term_states_fwd_probs']`
`.terminal_states`
`.terminal_states_probabilities`
`.terminal_states_memberships
Finish`

We call this semi-automatic terminal state identification as we manually pass the macrostates we would like to select, however, the actual cells belonging to each macrostate are assigned automatically. For initial states, everything works exactly the same, you can identify them fully automatically, or semi-automatically. Let’s compute the initial states fully automatically here:
g.predict_initial_states()
g.plot_macrostates(which="initial", s=100)
INFO Adding `adata.obs['init_states_fwd']`
`adata.obs['init_states_fwd_probs']`
`.initial_states`
`.initial_states_probabilities`
`.initial_states_memberships
Finish`

How can we check whether this is really the correct initial state? In this case, we have prior knowledge that we can use: we know that the initial state should be high for a number of Ductal cell markers. So let’s use these markers to compute a score that we can visualize across macrostates to confirm that we have the correct one.
# compute a score in scanpy by aggregating across a few ductal markers
sc.tl.score_genes(
adata, gene_list=["Bicc1", "Sox9", "Anxa2"], score_name="initial_score"
)
# write macrostates to AnnData
adata.obs["macrostates"] = g.macrostates
adata.uns["macrostates_colors"] = g.macrostates_memberships.colors
# visualize
ax = sc.pl.violin(
adata, keys="initial_score", groupby="macrostates", rotation=90, show=False
)
ax.legend(bbox_to_anchor=(1.04, 1), loc="upper left", borderaxespad=0)
plt.tight_layout()
plt.show()

In fact, the 'Ngn3 low EP_1' state scores highest, confirming the identification of this state as as initial state. The same strategy can be used to confirm terminal state identification, or to guide semi-automatic identification in the first place.
Closing matters¶
What’s next?¶
In this tutorial, you learned how to use CellRank’s GPCCA estimator to compute initial and terminal states of cellular dynamics. For the next steps, we recommend to:
go through the fate probabilities & driver genes tutorial to learn how to quantify cellular fate commitment.
take a look at the
APIto learn about parameter values you can use to adapt these computations to your data.read the original GPCCA publications to get familiar with the maths and ideas behind these computations [Reuter et al., 2019, Reuter et al., 2018].
Package versions¶
session_info2.session_info()